Mouse Cerebellum (Slideseq-V2)
[2]:
import numpy as np
import pandas as pd
import pathlib
import matplotlib.pyplot as plt
import matplotlib as mpl
import seaborn as sns
import scanpy as sc
import sys
sys.path.append('../src')
from utils import *
import pickle
import warnings
warnings.filterwarnings('ignore')
Preprocessing scRNA-ref
Ideally, the scRNAseq reference should from the exact same tissue that contains all the celltypes you expect to find in ST spots.
We will use a Mouse cerebellum scRNA-seq as reference dataset, the raw dataset in h5ad format (scRefSubsampled1000_cerebellum_singlenucleus.h5ad) is available in here.
Conveniently, we provided the pre-pocessed scRNA-ref (Ckpts_scRefs/Cerebellum/Ref_snRNA_cerebellum_qc.h5ad) as well as other relevent materials involved in the following example in here, so you can skip this part
[3]:
ad_sc = sc.read('scRefSubsampled1000_cerebellum_singlenucleus.h5ad')
ad_sc.obs['cell_type'] = ad_sc.obs['liger_ident'].map(lambda x:x.split('_')[-1])
cell_type_column = 'cell_type'
sc.pp.filter_cells(ad_sc, min_counts=500)
sc.pp.filter_cells(ad_sc, max_counts=20000)
sc.pp.filter_genes(ad_sc, min_cells=10)
ad_sc = ad_sc[:,~np.array([_.startswith('MT-') for _ in ad_sc.var.index])]
ad_sc = ad_sc[:,~np.array([_.startswith('mt-') for _ in ad_sc.var.index])]
ad_sc.X.max(),ad_sc.shape
[3]:
(7175.0, (14748, 20179))
[4]:
if ad_sc.X.max()<20:
ad_sc.X = np.exp(ad_sc.X)-1
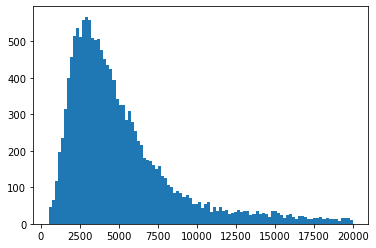
plt.hist(ad_sc.X.sum(1),bins=100)
plt.show()
[5]:
ad_sc.raw = ad_sc.copy()
sc.pp.normalize_total(ad_sc, inplace=True, target_sum=5e3)
sc.pp.highly_variable_genes(ad_sc, flavor='seurat_v3',n_top_genes=1000)
sc.tl.rank_genes_groups(ad_sc, groupby=cell_type_column, method='wilcoxon')
markers_df = pd.DataFrame(ad_sc.uns["rank_genes_groups"]["names"]).iloc[0:50, :]
markers = list(np.unique(markers_df.melt().value.values))
markers = list(set(ad_sc.var.loc[ad_sc.var['highly_variable']==1].index)|set(markers)) # highly variable genes + cell type marker genes
ligand_recept = list(set(pd.read_csv('../extdata/ligand_receptors.txt',sep='\t').melt()['value'].values))
# if scRNA-seq reference is from human tissue, run following code to make gene name consistent
# ligand_recept = [_.upper() for _ in ligand_recept]
add_genes = ['Cadm3','Ptprt','Aldoc','Gria1'] # some important genes that we interested
markers = markers+add_genes+ligand_recept
len(markers)
WARNING: It seems you use rank_genes_groups on the raw count data. Please logarithmize your data before calling rank_genes_groups.
[5]:
2217
[6]:
ad_sc.var.loc[ad_sc.var.index.isin(markers),'Marker'] = True
ad_sc.var['Marker'] = ad_sc.var['Marker'].fillna(False)
ad_sc.var['highly_variable'] = ad_sc.var['Marker']
sc.pp.log1p(ad_sc)
sc.pp.pca(ad_sc,svd_solver='arpack', n_comps=30, use_highly_variable=True)
ad_sc.X.max()
[6]:
7.6586394
[7]:
sc.pp.neighbors(ad_sc, metric='cosine', n_neighbors=30, n_pcs = 30)
sc.tl.umap(ad_sc, min_dist = 0.5, spread = 1, maxiter=60)
[8]:
fig, axs = plt.subplots(1, 1, figsize=(10, 10))
sc.pl.umap(
ad_sc, color=cell_type_column, size=15, frameon=False, show=False, ax=axs,legend_loc='on data'
)
plt.tight_layout()

[9]:
#ad_sc.write('../Ckpts_scRefs/Cerebellum/Ref_snRNA_cerebellum_qc.h5ad')
[ ]:
Learning the gene expression distribution of scRNA-seq reference using score-based model
We use four GPUs to train scRNA-seq reference in parallel.
python ./src/Train_scRef.py \
--ckpt_path ./Ckpts_scRefs/Cerebellum \
--scRef ./Ckpts_scRefs/Cerebellum/Ref_snRNA_cerebellum_qc.h5ad \
--cell_class_column cell_type \
--gpus 0,1,2,3
The checkpoints and sampled psuedo-cells will be saved in ./Ckpts_scRefs/Cerebellum, e.g, model_5000.pt, model_5000.h5ad
Conveniently, we provided the pre-trained checkpoint (Ckpts_scRefs/Cerebellum/model_5000.pt) in here, so you can skip this part
[ ]:
[ ]:
Run SpatialScope
Step1: Singlet Doublet Classification
Because Slide-seq data lacks paired histological image, we replace the first step ”Nuclei segmentation” with ”Singlet/Doublet classification” inspired by RCTD. Of note, this step is also applicable to other high resolution ST data such as HDST, DBiT-seq etc.
Step2: Cell type identification
Step 3: Gene expression decomposition
set -ex;
declare -a arr=(“0,1000” “1000,2000” “2000,3000” “3000,4000” “4000,5000” “5000,6000” “6000,7000” “7000,8000” “8000,9000” “9000,10000” “10000,11000” “11000,11626”)
for i in “${arr[@]}”
do
done
[ ]:
Cell type identification result (Fig. a)
[114]:
ad_sp = sc.read('../output/cere/sp_adata.h5ad')
ad_sc = sc.read('../Ckpts_scRefs/Cerebellum/Ref_snRNA_cerebellum_qc.h5ad')
[110]:
ad_sp.uns['cell_locations'].head()
[110]:
| spot_index | spot_class | first_type | second_type | min_score | singlet_score | first_type_weight | second_type_weight | x | y | cell_nums | cell_index | cell_index_n | cell_type | discrete_label | discrete_label_ct | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | GCCTTGCTGAGCTC | singlet | Bergmann | Granule | 445.194605 | 463.437230 | 0.824780 | 0.175220 | 1632.575000 | 3463.375000 | 1 | GCCTTGCTGAGCTC_0 | 0 | Bergmann | 1 | Bergmann |
| 1 | GGTTCGCGACCACA | singlet | Granule | Ependymal | 237.770038 | 252.347030 | 0.745520 | 0.254480 | 3078.088889 | 1795.177778 | 1 | GGTTCGCGACCACA_0 | 0 | Granule | 9 | Granule |
| 2 | CAGAGGCTTTTTTT | singlet | Astrocytes | Granule | 378.295186 | 383.238369 | 0.882583 | 0.117417 | 4682.637363 | 2342.197802 | 1 | CAGAGGCTTTTTTT_0 | 0 | Astrocytes | 0 | Astrocytes |
| 3 | CTTCGGATTTTTTT | singlet | Oligodendrocytes | Astrocytes | 327.364298 | 327.304644 | 0.998508 | 0.001492 | 4178.563830 | 2645.021277 | 1 | CTTCGGATTTTTTT_0 | 0 | Oligodendrocytes | 15 | Oligodendrocytes |
| 4 | TGTGGGGAATTTTT | singlet | Granule | Bergmann | 207.347290 | 208.740884 | 0.950068 | 0.049932 | 2757.945652 | 1932.380435 | 1 | TGTGGGGAATTTTT_0 | 0 | Granule | 9 | Granule |
[108]:
color_dict = {'Astrocytes': '#1f77b4',
'Bergmann': '#ff7f0e',
'Granule': '#2ca02c',
'Purkinje': '#d62728',
'MLI2': '#9467bd',
'MLI1': '#8c564b',
'Oligodendrocytes': '#e377c2',
'Lugaro': '#7f7f7f',
'Polydendrocytes': '#bcbd22',
'Golgi': '#17becf',
'Candelabrum': '#98df8a',
'Globular': '#f7b6d2',
'Microglia': '#aec7e8',
'Macrophages': '#dbdb8d',
'Choroid': '#c7c7c7',
'UBCs': '#c49c94',
'Ependymal': '#9edae5',
'Fibroblast': '#ff9896',
'Endothelial': '#ffbb78'}
[111]:
plt.subplots(figsize=(10,8.5))
sns.scatterplot(data=ad_sp.uns['cell_locations'], x="x", y="y",s=10,hue='discrete_label_ct',palette=color_dict,hue_order=np.unique(res_p2['cell_type']),legend=True)#rocket_r
plt.axis('off')
plt.legend(bbox_to_anchor=(0.97, .98),framealpha=0)
plt.show()

[134]:
ad_sp.shape
[134]:
(11626, 2312)
[ ]:
Dropouts correction (Fig. d)
[140]:
# We start by concatenating all the generated_cells_spotX_X.h5ad together
ad_scst = ConcatCells(s=0,e=12000,inter=1000,es=11626,file_path='../output/cere/',prefix='generated_cells_spot',suffix='.h5ad')
[189]:
sc.pp.log1p(ad_scst)
sc.pp.log1p(ad_sp)
[192]:
gene = 'Ninj2'
VisualDE(ad_sp,gene,0)

[195]:
VisualDE(ad_scst,gene,.01)

[173]:
VisualscDE(ad_sc,gene)

[182]:
gene = 'Foxp4'
VisualDE(ad_sp,gene,0)
[181]:
VisualDE(ad_scst,gene,0.01)

[183]:
VisualscDE(ad_sc,gene)

[155]:
fig, axs = plt.subplots(1, 1, figsize=(10, 10))
sc.pl.umap(
ad_sc, color="cell_type", size=10, frameon=False, show=False, ax=axs,legend_loc='on data',palette=color_dict
)
plt.tight_layout()

[ ]:
Cellular communications detection
refer to cell-cell interaction analysis in Human heart and Human-Heart-CCell-interaction.ipynb
Representative examples (Fig. f)
[196]:
L_gene,R_gene,L_ct,R_ct = 'Apoe','Sorl1','MLI2','Purkinje'
fig, ax = plt.subplots(1, 1, figsize=(10.2, 8),dpi=100,facecolor='#fafafa')
PlotLRGenes(ad_scst,L_gene,R_gene,L_ct,R_ct,title='${}$'.format(L_gene+'-'+R_gene),ax=ax,invertY=False,s=10)

[198]:
L_gene,R_gene,L_ct,R_ct = 'Mdk','Ptprz1','Bergmann','Granule'
fig, ax = plt.subplots(1, 1, figsize=(10.2, 8),dpi=100,facecolor='#fafafa')
PlotLRGenes(ad_scst,L_gene,R_gene,L_ct,R_ct,title='${}$'.format(L_gene+'-'+R_gene),ax=ax,invertY=False,s=20)

[199]:
L_gene,R_gene,L_ct,R_ct = 'Nlgn2','Nrxn3','Granule','Purkinje'
fig, ax = plt.subplots(1, 1, figsize=(10.2, 8),dpi=100,facecolor='#fafafa')
PlotLRGenes(ad_scst,L_gene,R_gene,L_ct,R_ct,title='${}$'.format(L_gene+'-'+R_gene),ax=ax,invertY=False,s=20)

[ ]:
[ ]:
Cellular communications only detected in the corrected Slide-seq data by SpatialScope (Fig. g)
[208]:
with open('../output/cere/InitProp.pickle', 'rb') as handle:
InitProp = pickle.load(handle)
[212]:
ad_sp.obs = ad_sp.obs.merge(InitProp['results']['results_df'][['spot_class','first_type','second_type']],left_index=True,right_index=True,how='left')
[215]:
ad_sp.obs['discrete_label_ct'] = ad_sp.obs['first_type']
[224]:
sns.set_context('paper',font_scale=3.5)
L_gene,R_gene,L_ct,R_ct = 'Psap','Gpr37l1','MLI1','Astrocytes'
fig, ax = plt.subplots(1, 1, figsize=(10.2, 8),dpi=100,facecolor='white')
PlotLRGenes(ad_sp,L_gene,R_gene,L_ct,R_ct,title='${}$'.format(L_gene+'-'+R_gene),ax=ax,invertY=False,s=30,perc=0.01)

[225]:
sns.set_context('paper',font_scale=3.5)
L_gene,R_gene,L_ct,R_ct = 'Psap','Gpr37l1','MLI1','Astrocytes'
fig, ax = plt.subplots(1, 1, figsize=(10.2, 8),dpi=100,facecolor='white')
PlotLRGenes(ad_scst,L_gene,R_gene,L_ct,R_ct,title='${}$'.format(L_gene+'-'+R_gene),ax=ax,invertY=False,s=30,perc=0.01)

[217]:
gene = L_gene
celltype = L_ct
sns.set_context('paper',font_scale=2.4)
fig, ax = plt.subplots(1, 1, figsize=(8, 6),dpi=100)
sns.kdeplot(data=ad_sp[ad_sp.obs['discrete_label_ct']==celltype,gene].to_df(), x=gene,ax=ax,fill=True,color='#D6D657',label='Slide-seq')
sns.kdeplot(data=ad_scst[ad_scst.obs['discrete_label_ct']==celltype,gene].to_df(), x=gene,ax=ax,fill=True,color='#BF8AD0',label='SpatialScope',clip=[-1,10])
ax.set_xlabel('${}$ expression in {}'.format(gene,celltype))
plt.legend(loc='best')
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
plt.show()

[218]:
gene = R_gene
celltype = R_ct
sns.set_context('paper',font_scale=2.4)
fig, ax = plt.subplots(1, 1, figsize=(8, 6),dpi=100)
sns.kdeplot(data=ad_sp[ad_sp.obs['discrete_label_ct']==celltype,gene].to_df(), x=gene,ax=ax,fill=True,color='#D6D657',label='Slide-seq')
sns.kdeplot(data=ad_scst[ad_scst.obs['discrete_label_ct']==celltype,gene].to_df(), x=gene,ax=ax,fill=True,color='#BF8AD0',label='SpatialScope',clip=[-1,10])
ax.set_xlabel('${}$ expression in {}'.format(gene,celltype))
#plt.legend(loc='best')
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
plt.show()

[222]:
sns.set_context('paper',font_scale=3.5)
L_gene,R_gene,L_ct,R_ct = 'Ptn','Ptprz1','Astrocytes','MLI2'
fig, ax = plt.subplots(1, 1, figsize=(10.2, 8),dpi=100,facecolor='#fafafa')
PlotLRGenes(ad_sp,L_gene,R_gene,L_ct,R_ct,title='${}$'.format(L_gene+'-'+R_gene),ax=ax,invertY=False,s=30)

[223]:
sns.set_context('paper',font_scale=3.5)
L_gene,R_gene,L_ct,R_ct = 'Ptn','Ptprz1','Astrocytes','MLI2'
fig, ax = plt.subplots(1, 1, figsize=(10.2, 8),dpi=100,facecolor='#fafafa')
PlotLRGenes(ad_scst,L_gene,R_gene,L_ct,R_ct,title='${}$'.format(L_gene+'-'+R_gene),ax=ax,invertY=False,s=30)

[220]:
gene = L_gene
celltype = L_ct
sns.set_context('paper',font_scale=2.4)
fig, ax = plt.subplots(1, 1, figsize=(8, 6),dpi=100)
sns.kdeplot(data=ad_sp[ad_sp.obs['discrete_label_ct']==celltype,gene].to_df(), x=gene,ax=ax,fill=True,color='#D6D657',label='Slide-seq')
sns.kdeplot(data=ad_scst[ad_scst.obs['discrete_label_ct']==celltype,gene].to_df(), x=gene,ax=ax,fill=True,color='#BF8AD0',label='SpatialScope',clip=[-1,10])
ax.set_xlabel('${}$ expression in {}'.format(gene,celltype))
plt.legend(loc='best')
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
plt.show()

[221]:
gene = R_gene
celltype = R_ct
sns.set_context('paper',font_scale=2.4)
fig, ax = plt.subplots(1, 1, figsize=(8, 6),dpi=100)
sns.kdeplot(data=ad_sp[ad_sp.obs['discrete_label_ct']==celltype,gene].to_df(), x=gene,ax=ax,fill=True,color='#D6D657',label='Slide-seq')
sns.kdeplot(data=ad_scst[ad_scst.obs['discrete_label_ct']==celltype,gene].to_df(), x=gene,ax=ax,fill=True,color='#BF8AD0',label='SpatialScope',clip=[-1,10])
ax.set_xlabel('${}$ expression in {}'.format(gene,celltype))
plt.legend(loc='best')
ax.spines['top'].set_visible(False)
ax.spines['right'].set_visible(False)
ax.set_ylabel('')
plt.show()

[ ]:
Comparison of cell counts between raw Slide-seq data and corrected Slide-seq data (Fig. i)
[226]:
cellcounts = pd.DataFrame(ad_scst.obs.discrete_label_ct.value_counts()).merge(pd.DataFrame(ad_sp.obs.discrete_label_ct.value_counts()),how='left',right_index=True,left_index=True)
cellcounts = cellcounts.fillna(0)
[227]:
cellcounts.head()
[227]:
| discrete_label_ct_x | discrete_label_ct_y | |
|---|---|---|
| Granule | 6241 | 5737 |
| Oligodendrocytes | 1042 | 930 |
| Purkinje | 975 | 750 |
| Bergmann | 715 | 1435 |
| Astrocytes | 698 | 1066 |
[228]:
sns.set_context('paper',font_scale=1.7)
width=0.4
fig, ax = plt.subplots(1, 1, figsize=(4, 16), dpi=100)
#plt.figure()
ax.margins(0.1, 0.01)
bars2 = ax.barh(np.arange(cellcounts.shape[0]-1)-width/2,cellcounts['discrete_label_ct_x'].values[1:],height=width,color='#BF8AD0',label='SpatialScope')
bars1 = ax.barh(np.arange(cellcounts.shape[0]-1)+width/2,cellcounts['discrete_label_ct_y'].values[1:],height=width,color='#D6D657',label='Slide-seq')
plt.yticks(np.arange(cellcounts.shape[0]-1))
ax.set_yticklabels(cellcounts.index.values[1:],rotation=30,ha='right')
plt.legend(loc=4)
#plt.ylabel('cell type')
plt.xlabel('cell counts')
ax.spines['bottom'].set_visible(False)
ax.spines['right'].set_visible(False)
ax.bar_label(bars1,fontsize=14,padding=5)
ax.bar_label(bars2,fontsize=14,padding=5)
ax.invert_yaxis()
ax.xaxis.tick_top()
ax.xaxis.set_label_position('top')
plt.show()

[ ]: